Objective & Goals
- The Education, Science Communication, and Society group focuses on theories, methods, and practices through which the ecologies of education and science communication are structured. With a special focus on chemical science, the research group recognizes the ubiquitous and polymorphic nature of digital media as a privileged means of redefining the configuration and comprehensiveness of these ecologies in a global and systemic society. The contextualized and relational explication of chemistry in relation to other disciplinary areas, knowledge, cultural and social contexts, are still an important focus of action and investigation.
The research interests of the group Education, Science Communication and Society are organized into four major areas
- Pre and post-graduate teacher education and the dissemination of effective pedagogical practices for science teaching and learning from the basic to higher level
- Models, processes, digital teaching-learning ecologies and modes of evaluation of technology integration in teaching and communication of science
- Models and processes of dissemination, diffusion, appropriation and re-mediation of scientific knowledge and citizen participation in science
- Nature of science and nature of religion in a school, and cultural context
Publications
Show All Group PublicationsEstimations of the thermodynamic properties of halogenated benzenes as they relate to their environment mobility
Team: João Monte; Ana Rita Almeida
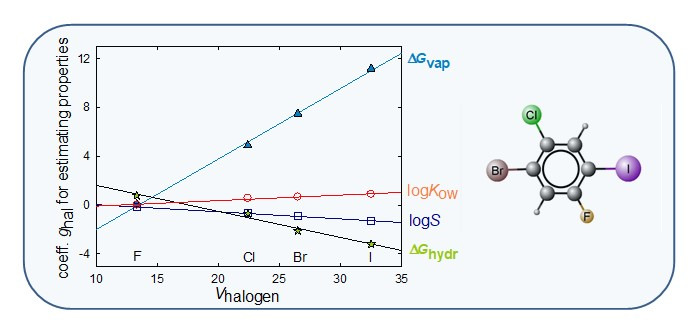
Halogenated benzenes is a wide group of aromatic compounds that contains virtually ca. 1500 distinct ones (Monte et al., 2015). Most of these compounds are potentially pollutants and so the knowledge of their mobility properties is required to predict their fate in the environment. These properties include volatility, aqueous solubility, partition coefficients and Henry’s law constants. Their values reflect the intensity of intermolecular forces that may be strongly affected by the existence of intramolecular interactions between neighbor substituent groups. For halogen atoms these intramolecular interactions seem to be negligible allowing the development of very simple predictive equations of the properties referred to above. It was also shown that the coefficients of those equations are well correlated with the volume of each halogen atom.
Chemosphere 2017 189, 590-598. link
Thermodynamic properties of 2,7-di-tert-butylfluorene -an experimental and computational study
Team: Juliana Oliveira, Vera Freitas, Rafael Notário, Maria das Dores Ribeiro da Silva, João Monte
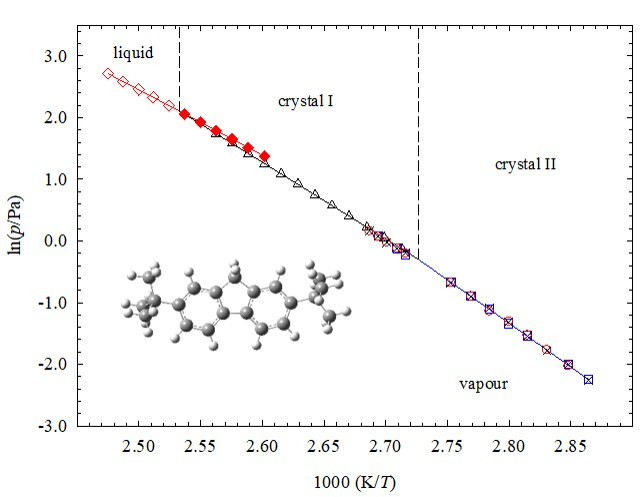
This work presents a comprehensive experimental and computational study of the thermodynamic properties of 2,7-di-tert-butylfluorene. The standard molar enthalpy of formation in the crystalline phase was derived from the standard molar energy of combustion, measured by static bomb combustion calorimetry. This property was also determined from a computational study at the G3(MP2)//B3LYP and G3 levels performed in this work. The enthalpies and temperatures of the crystal II - crystal (I) transition were determined from DSC experiments. The vapour pressures of the two crystalline and of the liquid phases (including supercooled liquid) were measured between (349.14 and 404.04) K, using two different experimental methods (static and Knudsen-effusion). From these results the standard molar enthalpies, entropies and Gibbs energies of sublimation and of vaporization were derived. The thermodynamic stability of the crystalline and gaseous phases was evaluated by the determination of their standard Gibbs energies of formation, at the temperature 298.15 K, and were critically compared with the ones reported in the literature for fluorene.
Journal of Chemical Thermodynamics 2016 101, 115-122. link
Experimental and Computational Thermochemical Study of Maleic Anhydride and Vinylene Carbonate
Team: Clara Sousa; M. Agostinha R. Matos; Victor M. F. Morais.
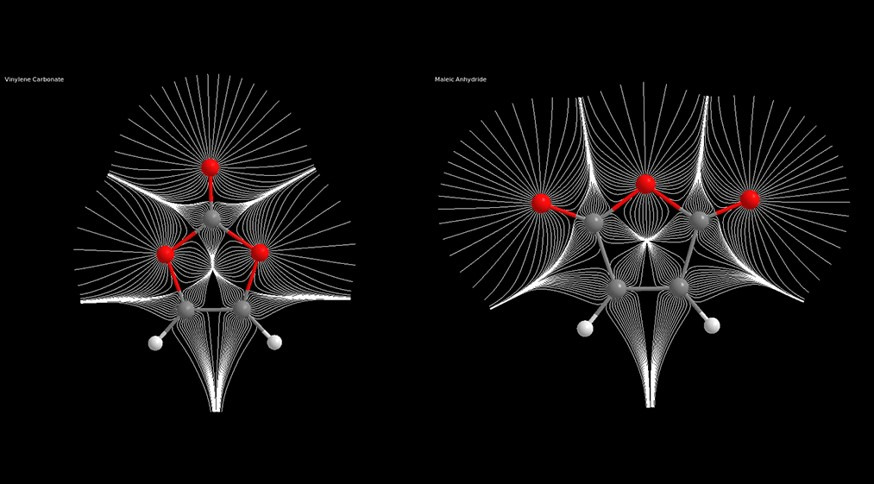
Accurate measurements of the standard enthalpy of combustion of vinylene carbonate and of maleic anhydride, after combination with their phase transition enthalpies , allowed the determination of the gas-phase enthalpies of formation. The enthalpy of formation for maleic anhydride results corroborate the previous reported values but with a considerably narrowing of the corresponding experimental error bar. On the contrary, for vinylene carbonate we obtained a very different value for the corresponding enthalpy of formation. Very accurate quantum chemical calculations on the title systems provided further assessment of our newly obtained experimental data. We have investigated the aromatic or antiaromatic behaviour for both systems using theoretical approaches ranging from the analysis of their magnetic properties (NICS), to the behaviour of the exchange-correlation function (Fermi hole) and to the energetic stabilization or unstabilization. In every case we were led to the conclusion that while vinylene carbonate can be viewed as an aromatic system, it revealed impossible to ascribe an unequivocally antiaromatic character to maleic anhydride, despite its ring π-electron counting indicates antiaromaticity
Journal of Physical Chemistry A, 2017 121, 9474-9484. link
Comprehensive Thermochemical Study of Cyclic Five and Six-membered N,N´-thioureas
Team: Ana Luísa Silva; Maria das Dores Ribeiro da Silva.
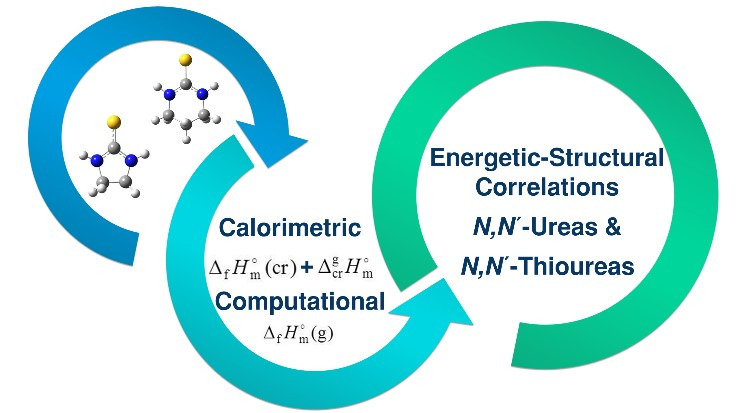
The assessment between the teratogenicity and the molecular structure of compounds related derivatives ethylenethiourea (imidazolidine-2-thione, ETU) has been studied, being performed modifications on the ring size and in the substituents, in order to evaluate the structural effects on the characteristics of the compounds. The results indicated a teratogenic activity and molecular structure correlation, closely linked with the imidazolidinethione structure and, although the interest of the energetic vs structural effects for the prediction of the chemical behaviour of ETU derivatives, such studies are scarce, being important to carry out them. Ethylenethiourea and N,N’-trimethylenethiourea, whose structures contain a thioamide group, are well known as ligands for the synthesis of coordination compounds, appearing in the literature extensively studied the inherent metal–biological species interactions.
An experimental and computational study of thermochemical and structural properties of ethylenethiourea (ETU) has been carried out. The enthalpies of combustion and sublimation, measured respectively by rotating-bomb combustion calorimetry and Calvet microcalorimetry, yielded the gas-phase enthalpy of formation of ETU, at T = 298.15 K. This latter parameter was also derived from high-level molecular orbital calculations at the G3(MP2)//B3LYP level of theory, leading to a value in excellent agreement with that one obtained from experimental data. With the purpose of evaluating the influence of the ring size in the enthalpy of formation of cyclic N,N´-thioureas derivatives, the calculation of the enthalpy of formation of N,N’-trimethylenethiourea (MTU) was performed using also the G3(MP2)//B3LYP approach. The effects of substituents (carbonyl and thiocarbonyl) on the molecular stability of several N-alkyl (cyclic) ureas/thioureas were also studied.
Journal of Chemical and Engineering Data 2017 62, 2584-2591. link
Research Group Members






RT2: Education, Science Communication, and Society
Research fellow / PhD student

RT2: Education, Science Communication, and Society
Team Researcher / RT5 - Group Leader

RT2: Education, Science Communication, and Society
Research fellow / PhD student
















RT2: Education, Science Communication, and Society
Collaborator / Reasearcher

RT2: Education, Science Communication, and Society
Team Researcher / Researcher



